

Chromatographie, auch bekannt als „chromatographische Analyse“, „Chromatographie“, ist eine Trenn- und Analysemethode, die in der analytischen Chemie, organischen Chemie, Biochemie und anderen Bereichen ein sehr breites Anwendungsspektrum hat.
Der Begründer der Chromatographie ist der russische Botaniker M. Tsvetter.Im Jahr 1906 veröffentlichte der russische Botaniker Zvetter die Ergebnisse seines Experiments: Um Pflanzenpigmente zu trennen, schüttete er Petroletherextrakt mit Pflanzenpigmenten in ein Glasröhrchen mit Calciumcarbonatpulver und eluierte es von oben nach unten mit Petrolether.Da unterschiedliche Pigmente unterschiedliche Adsorptionskapazitäten auf der Oberfläche von Calciumcarbonatpartikeln haben, bewegen sich unterschiedliche Pigmente beim Auslaugungsprozess mit unterschiedlicher Geschwindigkeit nach unten und bilden so Bänder unterschiedlicher Farbe.Die Pigmentbestandteile wurden getrennt.Er nannte diese Trennmethode Chromatographie.
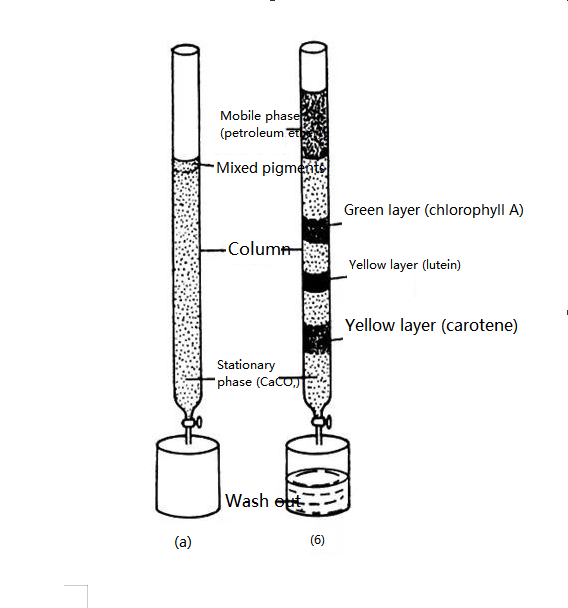
Schematische Darstellung eines Experiments zur Pigmenttrennung in Pflanzenblättern
Mit der kontinuierlichen Weiterentwicklung der Trennmethoden werden immer mehr farblose Substanzen zum Gegenstand der Trennung, auch die Chromatographie verliert nach und nach die Bedeutung von „Farbe“, der Name ist jedoch bis heute gebräuchlich.
Chromatographische Klassifizierung
Das Wesen der Chromatographie ist ein Prozess, bei dem die zu trennenden Moleküle zwischen der stationären Phase und der mobilen Phase aufgeteilt und ausgeglichen werden.Verschiedene Substanzen verteilen sich unterschiedlich auf die beiden Phasen, wodurch sie sich mit der mobilen Phase unterschiedlich schnell bewegen.Durch die Bewegung der mobilen Phase werden verschiedene Bestandteile der Mischung auf der stationären Phase voneinander getrennt.Je nach Mechanismus lässt es sich in verschiedene Kategorien einteilen.
1, gemäß der zweiphasigen Klassifizierung des physikalischen Zustands
Mobile Phase: Gaschromatographie, Flüssigkeitschromatographie, überkritische Flüssigkeitschromatographie
Stationäre Phase: Gas-Feststoff, Gas-Flüssigkeit;Flüssig-fest, flüssig-flüssig
2, entsprechend der Form der stationären Phasenklassifizierung
Säulenchromatographie: gepackte Säulenchromatographie, Kapillarsäulenchromatographie, mikrogepackte Säulenchromatographie, präparative Chromatographie
Ebenenchromatographie: Papierchromatographie, Dünnschichtchromatographie, Polymermembranchromatographie
3, klassifiziert nach dem Trennmechanismus
Adsorptionschromatographie: Verschiedene Komponenten werden nach ihrem Adsorptions- und Desorptionsvermögen an Adsorbentien getrennt
Verteilungschromatographie: Die verschiedenen Komponenten werden nach ihrer Löslichkeit im Lösungsmittel getrennt
Molekulare Ausschlusschromatographie: entsprechend der Größe der Molekülgröße der Trennung. ln Ionenaustauschchromatographie: verschiedene Komponenten der Affinität für die Ionenaustauschharztrennung
Affinitätschromatographie: Trennung anhand der Anwesenheit einer spezifischen Affinität zwischen biologischen Makromolekülen
Kapillarelektrophorese: Die Komponenten wurden nach Unterschieden in der Mobilität und/oder im Verteilungsverhalten getrennt
Chirale Chromatographie wird zur Trennung und Analyse chiraler Arzneimittel verwendet, die in drei Kategorien unterteilt werden können: chirale Derivatisierungsreagenzmethode;Additive Methode der chiralen mobilen Phase;Chirale stationäre Phasenauflösungsmethode
Grundlegende Terminologie für die Chromatographie
Die Kurven, die man durch Auftragen der Antwortsignale der Komponenten nach der Detektion der chromatographischen Trennung gegen die Zeit erhält, werden Chromatogramme genannt.
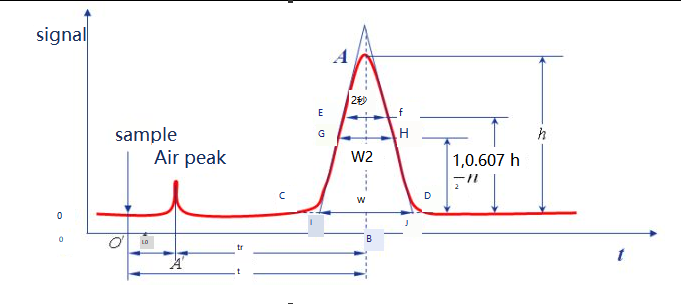
Grundlinie:Unter bestimmten chromatographischen Bedingungen wird die Kurve des Signals, das erzeugt wird, wenn nur die mobile Phase das Detektorsystem passiert, als Basislinie bezeichnet, wie in der ot-Linie dargestellt.Wenn die Versuchsbedingungen stabil waren, war die Basislinie eine Linie parallel zur horizontalen Achse.Die Basislinie spiegelt das Rauschen des Instruments, hauptsächlich des Detektors, über die Zeit wider.
Spitzenhöhe:der vertikale Abstand zwischen dem chromatographischen Peakpunkt und der Basislinie, bezeichnet mit h, wie in der AB'-Linie dargestellt.
Bereichsbreite:Die Bereichsbreite des chromatographischen Peaks steht in direktem Zusammenhang mit der Trenneffizienz.Es gibt drei Methoden zur Beschreibung der chromatographischen Peakbreite: Standardabweichung σ, Peakbreite W und FWHM W1/2.
Standardabweichung (σ):σ ist der halbe Abstand zwischen den beiden Wendepunkten auf der Normalverteilungskurve, und der Wert von σ gibt den Grad der Streuung der Komponenten von der Spalte weg an.Je größer der Wert von σ ist, desto stärker sind die Abwasserbestandteile verteilt und desto schlechter ist der Trenneffekt.Umgekehrt werden die Abwasserbestandteile konzentriert und die Trennwirkung ist gut.
Spitzenbreite W:Die Schnittpunkte auf beiden Seiten des chromatographischen Peaks werden als Tangentenlinien verwendet, und der Schnittpunkt auf der Basislinie wird Peakbreite oder Basislinienbreite genannt, die auch als W ausgedrückt werden kann, wie in Abbildung IJ dargestellt.Nach dem Prinzip der Normalverteilung kann nachgewiesen werden, dass die Beziehung zwischen Peakbreite und Standardabweichung W=4σ ist.
W1/2:Die Peakbreite bei halber Peakhöhe wird als FWHM bezeichnet, wie für den Abstand von GH gezeigt.W1/2=2,355σ, W=1,699W1/2.
W1/2 und W werden beide von σ abgeleitet und zusätzlich zur Messung des Säuleneffekts zur Berechnung von Peakflächen verwendet.Die FWHM-Messung ist bequemer und wird am häufigsten verwendet.
kurze Zusammenfassung
Aus der chromatographischen Peak-Outflow-Kurve lassen sich folgende Ziele erreichen:
a: Die qualitative Analyse wurde basierend auf dem Retentionswert der chromatographischen Peaks durchgeführt
b, quantitative Analyse basierend auf der Fläche oder dem Peak des chromatographischen Peaks
C. Die Trenneffizienz der Säule wurde anhand des Retentionswerts und der Peakbreite des chromatographischen Peaks bewertet
Die Berechnungsformel für die Chromatographie
1. Aufbewahrungswert
Der Retentionswert ist ein Parameter, der den Grad der Retention einer Probenkomponente in der Säule beschreibt und als Indikator für die chromatographische Charakterisierung dient.Seine Darstellungsmethode ist wie folgt:
Retentionszeit tR
Zeitpunkt des TodestM
Passen Sie die Retentionszeit tR an'=tR-tM
(Gesamtzeit in der stationären Phase verbracht)
Retentionsvolumen
VR=tR*F. (unabhängig von der Geschwindigkeit der mobilen Phase)
Totvolumen
VM=tM*Fc
(Der Raum, der nicht von der stationären Phase im Strömungsweg vom Injektor zum Detektor eingenommen wird)
Passen Sie das Retentionsvolumen VR an'=t'R*Fc
2. Relativer Retentionswert
Der relative Retentionswert, auch Trennfaktor, Verteilungskoeffizientenverhältnis oder relativer Kapazitätsfaktor genannt, ist das Verhältnis der angepassten Retentionszeit (Volumen) der getesteten Komponente zur angepassten Retentionszeit (Volumen) des Standards unter bestimmten chromatographischen Bedingungen.

Relative Retentionswerte wurden verwendet, um den Einfluss bestimmter Betriebsbedingungen wie Durchflussrate und Fixiermittelverlust auf die Retentionswerte zu eliminieren.Der Maßstab für den relativen Retentionswert kann ein Bestandteil der untersuchten Probe oder eine künstlich hinzugefügte Verbindung sein.
3. Aufbewahrungsindex
Der Retentionsindex ist der Retentionsindex der zu prüfenden Substanz i in einer festen Lösung X. Als Referenzsubstanzen werden zwei n-Alane ausgewählt, von denen eines die Kohlenstoffzahl N und das andere N+n hat.Ihre angepasste Retentionszeit beträgt t 'r (N) bzw. t 'r (N+n), so dass die angepasste Retentionszeit t 'r (i) der zu testenden Substanz i genau zwischen ihnen liegt, d. h. t 'r (N).
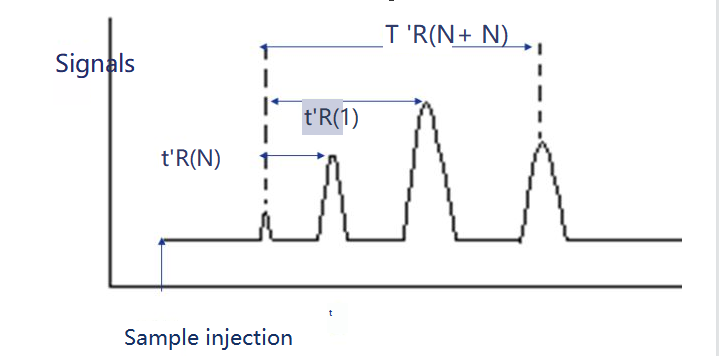
Der Retention-Index kann wie folgt berechnet werden.
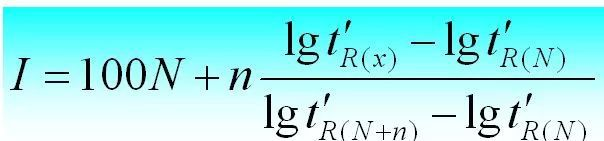
4. Kapazitätsfaktor (k)
Im Gleichgewicht das Verhältnis der Masse einer Komponente in der stationären Phase (s) zur mobilen Phase (m), genannt Kapazitätsfaktor.Die Formel lautet wie folgt:
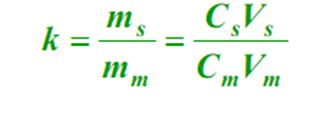
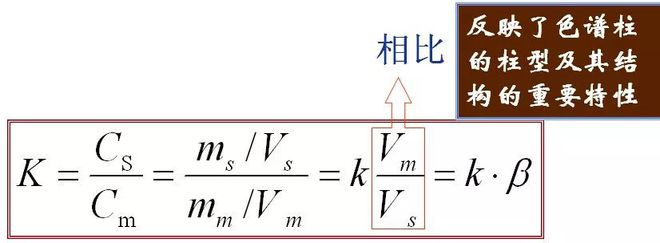
5、Verteilungskoeffizient (K) Im Gleichgewicht das Verhältnis der Konzentration einer Komponente in der stationären Phase (s) zur mobilen Phase (m), genannt Verteilungskoeffizient.Die Formel lautet wie folgt

Die Beziehung zwischen K und k:
Es spiegelt den Säulentyp und seinen Knoten als wichtige Struktureigenschaften wider
kurze Zusammenfassung
Zusammenhang zwischen Retentionswert und Kapazitätsfaktor und Verteilungskoeffizient:
Die chromatographische Trennung basiert auf dem Unterschied in der Adsorptions- oder Lösungsfähigkeit jeder Komponente in einer festen relativen Probe, der quantitativ durch die Größe des Werts des Verteilungskoeffizienten K (oder Kapazitätsfaktors k) ausgedrückt werden kann.
Die Komponenten mit starker Adsorptions- oder Lösungsfähigkeit haben einen großen Verteilungskoeffizienten (oder Kapazitätsfaktor) und eine lange Retentionszeit.Umgekehrt haben Komponenten mit schwacher Adsorption oder Löslichkeit einen kleinen Verteilungskoeffizienten und eine kurze Retentionszeit.
Grundlegende Theorie der Chromatographie
1. Tray-Theorie
(1) Vorgebracht – thermodynamische Theorie
Es begann mit dem von Martin und Synge vorgeschlagenen Turmplattenmodell.
Fraktionierungskolonne: Auf dem Boden wird das Gas-Flüssigkeits-Gleichgewicht je nach Siedepunkt der verschiedenen Trennungen mehrmals eingestellt.
Säule: Die Komponenten werden durch mehrere Verteilungen zwischen den beiden Phasen ausgeglichen und nach unterschiedlichen Verteilungskoeffizienten getrennt.
(2) Hypothese
(1) Es gibt viele Böden in der Kolonne, und die Komponenten können innerhalb des Bodenintervalls (d. h. der Höhe des Bodens) schnell das Verteilungsgleichgewicht erreichen.
(2) Die mobile Phase tritt nicht kontinuierlich, sondern pulsierend in die Säule ein, d. h. jeder Durchgang entspricht einem Säulenvolumen.
(3) Wenn die Probe zu jeder Säulenplatte gegeben wurde, konnte die Diffusion der Probe entlang der Säulenachse vernachlässigt werden.
(4) Der Verteilungskoeffizient ist auf allen Böden gleich, unabhängig von der Menge der Komponenten.Das heißt, der Verteilungskoeffizient ist auf jedem Taban konstant.
(3) Prinzip
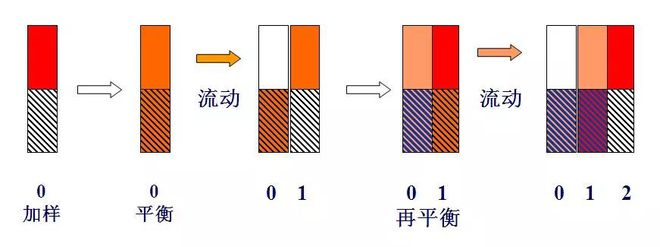
Schematische Darstellung der Tray-Theorie
Wenn eine Komponente mit einer Einheitsmasse, nämlich m = 1 (z. B. 1 mg oder 1 μg), zum Tablett Nr. 0 hinzugefügt wird und nach dem Verteilungsgleichgewicht, weil k = 1, nämlich ns = nm, nm = ns = 0,5.
Wenn ein Plattenvolumen (lΔV) Trägergas in Form einer Pulsation in Platte 0 eintritt, wird das Trägergas, das die nm-Komponente in der Gasphase enthält, auf Platte 1 gedrückt. Zu diesem Zeitpunkt befindet sich die ns-Komponente in der flüssigen Phase von Platte 0 und die nm-Komponente in der Gasphase von Platte 1 wird zwischen den beiden Phasen umverteilt.Daher beträgt die Gesamtmenge der in Platte 0 enthaltenen Komponenten 0,5, wobei die Gas- und Flüssigphase jeweils 0,25 beträgt, und die Gesamtmenge in Platte 1 beträgt ebenfalls 0,5.Die Gas- und Flüssigphasen betrugen ebenfalls 0,25.
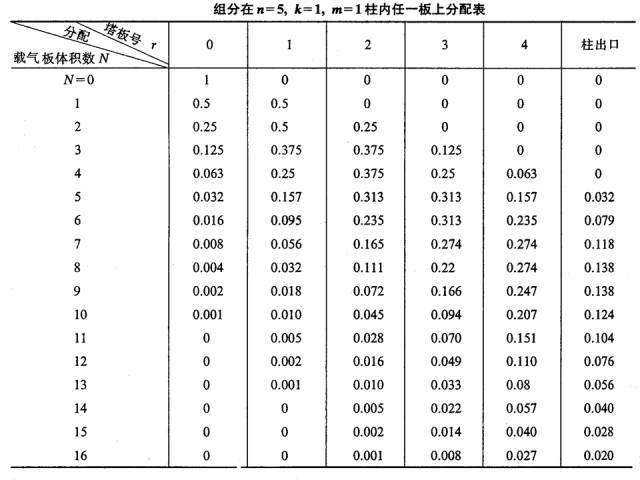
Dieser Vorgang wird jedes Mal wiederholt, wenn ein neues Plattenvolumen-Trägergas in die Säule gepulst wird (siehe Tabelle unten).
(4)Chromatographische Ausflusskurvengleichung

σ ist die Standardabweichung, ist die Retentionszeit, C ist die Konzentration zu jedem Zeitpunkt,
C ist die Injektionskonzentration, also die Gesamtmenge der Komponenten (Peakfläche A).
(5) Säuleneffizienzparameter
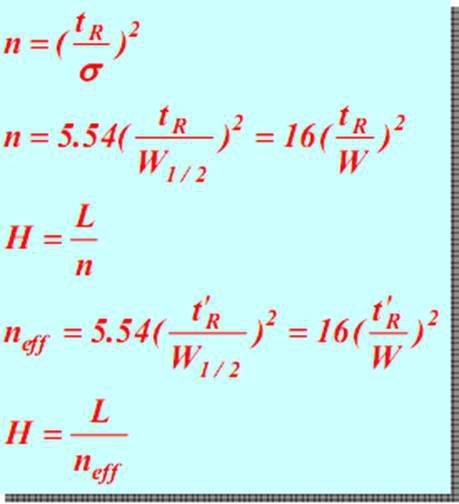
Bei einem konstanten tR gilt: Je kleiner W oder w 1/2 (d. h. je schmaler der Peak), desto größer ist die Anzahl der theoretischen Böden n, desto kleiner ist die theoretische Bodenhöhe und desto höher ist die Trenneffizienz der Säule.Dasselbe gilt auch für die Effektivtheorie von Tray Neff.Daher ist die theoretische Anzahl der Böden ein Index zur Bewertung der Effizienz von Kolonnen.
(5)Eigenschaften und Mängel
> Vorteile
Die Tray-Theorie ist semiempirisch und erklärt die Form der Abflusskurve
Die Aufteilungs- und Trennungsprozesse der Komponenten werden veranschaulicht
Es wird ein Index zur Bewertung der Effizienz der Kolonne vorgeschlagen
> Einschränkungen
Die Komponenten können das Verteilungsgleichgewicht in den beiden Phasen nicht wirklich erreichen:
Die Längsdiffusion von Komponenten in der Säule kann nicht ignoriert werden:
Der Einfluss verschiedener kinetischer Faktoren auf den Stoffübergangsprozess wurde nicht berücksichtigt.
Der Zusammenhang zwischen Säuleneffekt und Strömungsgeschwindigkeit der mobilen Phase kann nicht erklärt werden:
Es ist nicht klar, welche Hauptfaktoren den Säuleneffekt beeinflussen
Diese Probleme werden in der Ratentheorie zufriedenstellend gelöst.
2. Tariftheorie
Im Jahr 1956 stellten der niederländische Gelehrte VanDeemter et al.übernahm das Konzept der Tabletttheorie und kombinierte die kinetischen Faktoren, die die Höhe des Tabletts beeinflussen, stellte die kinetische Theorie des chromatographischen Prozesses vor – die Geschwindigkeitstheorie und leitete die VanDeemter-Gleichung ab.Der chromatographische Prozess wird als dynamischer Nichtgleichgewichtsprozess betrachtet und der Einfluss kinetischer Faktoren auf die Peakverbreiterung (Säuleneffekt) untersucht.
Später haben Giddings und Snyder et al.schlug die Geschwindigkeitsgleichung für die Flüssigkeitschromatographie (nämlich die Giddings-Gleichung) vor, die auf der VanDeemter-Gleichung (später als Geschwindigkeitsgleichung für die Gaschromatographie bezeichnet) und auf dem Eigenschaftsunterschied zwischen Flüssigkeit und Gas basiert.
(1) Van-Deemter-Gleichung
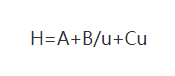
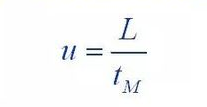
Wobei: H: die Höhe des Bretts ist
A: Koeffizient des Wirbeldiffusionsterms
B: Koeffizient des molekularen Diffusionsterms
C: Koeffizient des Stoffübergangswiderstandsterms
(2) Giddings-Gleichung
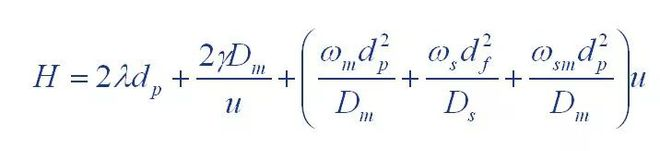
Quantitative und qualitative Analyse
(1) Qualitative Analyse
Bei der qualitativen chromatographischen Analyse geht es darum, die durch jeden chromatographischen Peak repräsentierten Verbindungen zu bestimmen.Da verschiedene Substanzen unter bestimmten chromatographischen Bedingungen bestimmte Retentionswerte aufweisen, kann der Retentionswert als qualitativer Index verwendet werden.Verschiedene chromatographische qualitative Methoden basieren derzeit auf Retentionswerten.
Allerdings können unterschiedliche Substanzen unter gleichen chromatographischen Bedingungen ähnliche oder gleiche Retentionswerte aufweisen, d. h. die Retentionswerte sind nicht exklusiv.Daher ist es schwierig, eine völlig unbekannte Probe allein anhand der Retentionswerte zu charakterisieren.Wenn auf der Grundlage des Verständnisses der Quelle, Art und des Zwecks der Probe eine vorläufige Beurteilung der Zusammensetzung der Probe getroffen werden kann, können die folgenden Methoden verwendet werden, um die durch den chromatographischen Peak dargestellte Verbindung zu bestimmen.
1. Qualitative Kontrolle mit reinen Substanzen
Unter bestimmten chromatographischen Bedingungen hat ein Unbekannter nur eine definierte Retentionszeit.Daher kann das Unbekannte qualitativ identifiziert werden, indem die Retentionszeit der bekannten reinen Substanz unter denselben chromatographischen Bedingungen mit der Retentionszeit der unbekannten Substanz verglichen wird.Wenn die beiden gleich sind, kann es sich bei der unbekannten Substanz um eine bekannte reine Substanz handeln;Ansonsten ist das Unbekannte nicht die reine Substanz.
Die Reinstoffkontrollmethode ist nur auf unbekannte Stoffe anwendbar, deren Zusammensetzung bekannt ist, deren Zusammensetzung relativ einfach ist und deren Reinstoff bekannt ist.
2. Methode des relativen Retentionswerts
Der relative Retentionswert α bezieht sich auf die Anpassung zwischen Komponente i und Referenzmaterialien. Verhältnis der Retentionswerte:
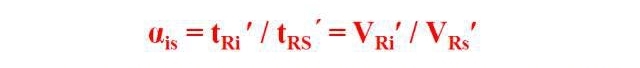
Sie ändert sich nur mit der Änderung des Fixiermittels und der Säulentemperatur und hat nichts mit anderen Betriebsbedingungen zu tun.
Bei einer bestimmten stationären Phase und Säulentemperatur werden die eingestellten Retentionswerte der Komponente i bzw. der Referenzsubstanz s gemessen und anschließend nach obiger Formel berechnet.Die erhaltenen relativen Retentionswerte können qualitativ mit den entsprechenden Werten in der Literatur verglichen werden.
3. Hinzufügen bekannter Substanzen zur Erhöhung der Peakhöhenmethode
Wenn die unbekannte Probe viele Komponenten enthält, sind die erhaltenen chromatographischen Peaks zu dicht, um mit der oben genannten Methode leicht identifiziert zu werden, oder wenn die unbekannte Probe nur für die Analyse des angegebenen Elements verwendet wird.
„Zuerst wird ein Chromatogramm einer unbekannten Probe erstellt, und dann wird ein weiteres Chromatogramm erhalten, indem man der unbekannten Probe eine bekannte Substanz hinzufügt.“Für solche Stoffe können Komponenten mit erhöhten Peakhöhen bekannt sein.
4. Behalten Sie die qualitative Methode des Index bei
Der Retentionsindex stellt das Retentionsverhalten von Substanzen auf Fixiermitteln dar und ist derzeit der am weitesten verbreitete und international anerkannte qualitative Index in der GC.Es bietet die Vorteile einer guten Reproduzierbarkeit, eines einheitlichen Standards und eines kleinen Temperaturkoeffizienten.
Der Retentionsindex hängt nur von den Eigenschaften der stationären Phase und der Säulentemperatur ab, nicht jedoch von anderen Versuchsbedingungen.Seine Genauigkeit und Reproduzierbarkeit sind ausgezeichnet.Solange die Säulentemperatur mit der der stationären Phase übereinstimmt, kann der Literaturwert zur Identifizierung herangezogen werden und es ist nicht notwendig, das reine Material zum Vergleich heranzuziehen.
(2)Quantitative Analyse
Grundlage für die chromatographische Quantifizierung:
Die Aufgabe der quantitativen Analyse besteht darin, die hundert Komponenten in der gemischten Probe zu finden
Bruchteil des Inhalts.Die chromatographische Quantifizierung basierte auf Folgendem: Bei konsistenten Betriebsbedingungen war
Die Masse (oder Konzentration) der gemessenen Komponente wird durch das vom Detektor ausgegebene Antwortsignal bestimmt
Es ist proportional.Nämlich:

Grundlage für die chromatographische Quantifizierung:
Die Aufgabe der quantitativen Analyse besteht darin, die hundert Komponenten in der gemischten Probe zu finden
Bruchteil des Inhalts.Die chromatographische Quantifizierung basierte auf Folgendem: Bei konsistenten Betriebsbedingungen war
Die Masse (oder Konzentration) der gemessenen Komponente wird durch das vom Detektor ausgegebene Antwortsignal bestimmt
Es ist proportional.Nämlich:
1. Methode zur Messung der Peakfläche
Bei der Peakfläche handelt es sich um die grundlegenden quantitativen Daten, die durch Chromatogramme bereitgestellt werden. Die Genauigkeit der Messung der Peakfläche wirkt sich direkt auf die quantitativen Ergebnisse aus.Für chromatographische Peaks mit unterschiedlichen Peakformen wurden unterschiedliche Messmethoden verwendet.
Es ist schwierig, den genauen Wert des Winters in einer quantitativen Analyse zu ermitteln:
Einerseits aufgrund der Schwierigkeit, das absolute Injektionsvolumen genau zu messen, andererseits
Die Peakfläche hängt von den chromatographischen Bedingungen ab und der Chromatographiestreifen sollte bei der Messung des Wertes aufbewahrt werden
Es ist weder möglich noch bequem, dasselbe zu tun.Und selbst wenn Sie es richtig machen können
Der genaue Wert, auch weil es keinen einheitlichen Standard gibt und nicht direkt angewendet werden kann.
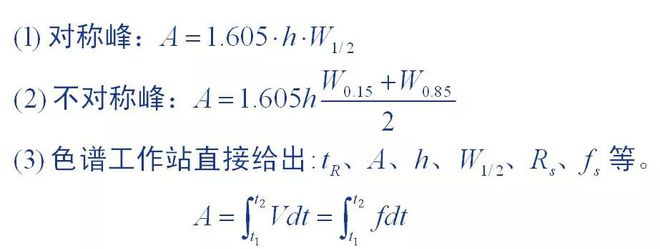
2. Quantitativer Korrekturfaktor
Definition des quantitativen Korrekturfaktors: Menge der in den Detektor gelangenden Komponenten (m)
Das Verhältnis seiner chromatographischen Peakfläche (A) oder Peakhöhe () ist eine Proportionalitätskonstante (,
Die Proportionalitätskonstante wird als absoluter Korrekturfaktor für die Komponente bezeichnet.
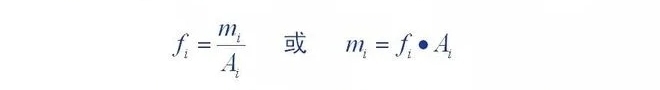
Es ist schwierig, den genauen Wert des Winters in einer quantitativen Analyse zu ermitteln:
Einerseits aufgrund der Schwierigkeit, das absolute Injektionsvolumen genau zu messen, andererseits
Die Peakfläche hängt von den chromatographischen Bedingungen ab und der Chromatographiestreifen sollte bei der Messung des Wertes aufbewahrt werden
Es ist weder möglich noch bequem, dasselbe zu tun.Und selbst wenn Sie es richtig machen können
Der genaue Wert, auch weil es keinen einheitlichen Standard gibt und nicht direkt angewendet werden kann.
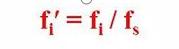
Das heißt, der relative Korrekturfaktor einer Komponente ist die Komponente und das Referenzmaterial s
Das Verhältnis der absoluten Korrekturfaktoren.
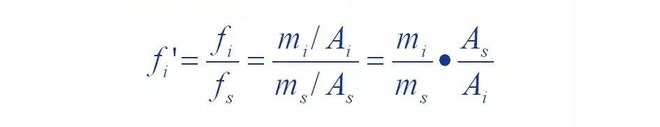
Es ist ersichtlich, dass sich der relative Korrekturfaktor aus der Qualität der Komponente im Vergleich zum Standard ergibt.
Wenn die Substanz s gleich ist, ist die Peakfläche des Referenzmaterials die Peakfläche der Komponente
Mehrere.Wenn eine Komponente die Masse m und die Peakfläche A hat, dann ist die Zahl f'A
Die Werte entsprechen der Peakfläche des Referenzmaterials mit einer Masse von.Mit anderen Worten,
Durch den relativen Korrekturfaktor können die Peakbereiche jeder Komponente getrennt werden
Umgerechnet auf die Peakfläche des Referenzmaterials gleich seiner Masse ergibt sich dann das Verhältnis
Der Standard ist einheitlich.Dies ist also die normalisierte Methode, um den Prozentsatz jeder Komponente zu ermitteln
Die Basis der Menge.
Methode zur Ermittlung des relativen Korrekturfaktors: Die relativen Korrekturfaktorwerte wurden nur mit denen verglichen
Die Messung hängt von der Norm und dem Meldertyp ab, nicht jedoch vom Betriebsstreifen
Es spielt keine Rolle.Daher können Werte aus Referenzen in der Literatur abgerufen werden.Wenn der Text
Sollten Sie im Angebot nicht den gewünschten Wert finden, können Sie ihn auch selbst ermitteln.Bestimmungsmethode
Methode: Eine bestimmte Menge der zu messenden Substanz aus zehn ausgewählten Referenzmaterialien → auf eine bestimmte Konzentration gebracht
Die chromatographischen Peakflächen A und As der beiden Komponenten wurden gemessen.
Das ist die Formel.
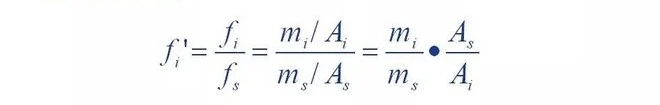
3. Quantitative Berechnungsmethode
(1) Flächennormalisierungsmethode
Zur Quantifizierung wurde die Summe des Gehalts aller Peak-freien Fraktionen zu 100 % berechnet
Die Methode heißt Normalisierung.Die Berechnungsformel lautet wie folgt:
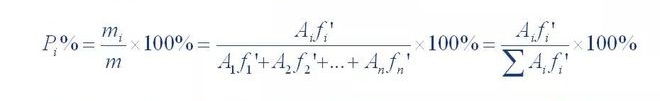
Wobei P,% der prozentuale Gehalt der getesteten Komponenten ist;A1, A2... A n ist Komponente 1. Die Peakfläche von 1~n;f'1, f'2... f'n ist der relative Korrekturfaktor für die Komponenten 1 bis n.
(2) externe Standardmethode
Die Methode des quantitativen Vergleichs zwischen dem Antwortsignal der zu testenden Komponente in der Probe und der reinen zu testenden Komponente als Kontrolle.
(3) Interne Standardmethode
Bei der sogenannten internen Standardmethode handelt es sich um eine Methode, bei der der Standardlösung des untersuchten Stoffes und der Probenlösung eine bestimmte Menge Reinstoff als interner Standard zugesetzt und anschließend analysiert und bestimmt wird.
(3)Standardadditionsmethode
Bei der Standardadditionsmethode, auch interne Additionsmethode genannt, wird eine bestimmte Menge (△C) hinzugefügt.
Die Referenz der Testsubstanz wurde der zu testenden Probenlösung hinzugefügt und der Test wurde dem Test hinzugefügt
Der Peak der Probenlösung nach der Substanz war höher als der der ursprünglichen Probenlösung
Der Flächenzuwachs (△A) wurde zur Berechnung der Konzentration der Substanz in der Probenlösung verwendet
Inhalt (Cx)
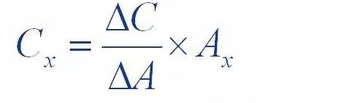
Dabei ist Ax die Peakfläche der zu messenden Substanz in der Originalprobe.

Zeitpunkt der Veröffentlichung: 27. März 2023